
دانستنی ها درباره بیماری تالاسمی
تالاسمی یک بیماری ژنتیکی است که نوع ماژور آن با اختلالات گسترده ای همراه است، برای داشتن فرزند سالم انجام مشاوره ژنتیک و آزمایشهای ژنتیکی قبل از بارداری ضروری است که میتواند در پیشگیری از تولد نوزاد مبتلا به تالاسمی و سایر بیماریهای ژنتیکی موثر باشد، به همین منظور باید حتما به یک آزمایشگاه ژنتیک مجهز در شهر تهران مراجعه کنید. بیماری تالاسمی شایعترین اختلال ژنتیکی تک ژنی با توارث اتوزومی مغلوب میباشد که از والدین به فرزندان منتقل میشود. تالاسمی ناشی از جهش در DNA ژنهایی است که هموگلوبین تولید میکنند و سبب کاهش تولید هموگلوبین در بدن میشود. هموگلوبین پروتئینی در گلبولهای قرمز خون میباشد که اکسیژن را از ریهها به بخشهای مختلف بدن حمل میکند. دو نوع اصلی تالاسمی وجود دارد، تالاسمی آلفا و تالاسمی بتا. شدت تالاسمی آلفا و بتا بستگی به این دارد که چه تعداد از چهار ژن برای آلفا گلوبین یا دو ژن برای بتاگلوبین وجود ندارد. تالاسمی اولین بار در سال 1925 توسط یک پزشک متخصص کودکان به نام Thomas B.cooly در شهر دیترویت آمریکا شناسایی شد. تالاسمی یک واژه یونانی است که ابتدا به صورت Thalassic anaemia و سپس به صورت Thalassaemia در آمد که از دو کلمه (Thalassa) به معنی دریا و (Emia) به معنی خون تشکیل شده است .هر چند این بیماری در سراسر جهان و در تمامی نژادها دیده می شود ولی شیوع آن در نواحی مدیترانه و خاورمیانه و شبه قاره هند و غرب آفریقا و جنوب شرقی آسیا بیشتر است، که این نواحی را کمربند تالاسمی می نامند. افراد مبتلا به تالاسمی به دلیل خونسازی غیر مؤثر و همولیزگلبول های قرمز به کم خونی مبتلا می شوند. در سال های اخیر آزمایش های قبل از ازدواج زوجین و تشخیص پیش از تولد (PND)بسیار کم هزینه تر از درمان و مراقبت از بیماران تالاسمی بوده و موجب کاهش این بیماری شده است. امروزه با آزمایشات هماتولوژیک و بررسی میزان هموگلوبینA2 از طریق الکتروفورز اکثر افراد ناقل این بیماری را مشخص می کند. طبقه بندی تالاسمی دو نوع طبقه بندی برای تالاسمی ارائه شده است: الف: طبقه بندی بالینی ب: طبقه بندی ژنتیکی طبقه بندی بالینی این بیماری به صورت شدید (ماژور) و خفیف (مینور) ظاهر میشود . اگر هر دو والدین دارای ژن معیوب باشند به صورت شدید یعنی ماژور و اگر یکی از والدین فقط ژن معیوب داشته باشد به صورت خفیف یعنی مینور ظاهر میشود. تالاسمی مینور تالاسمی مینور حالتی است که در آن شخص ، ناقل ژن تالاسمی است و می تواند آن را به نسل های بعد انتقال دهد ، در این حالت تغییری جزئی در خون فرد پدید می آید، بدین صورت که گلبولهای قرمز کمی کوچکتر از حالت عادی هستند اما به مقدار کافی وجود دارند و به طور طبیعی کار می کنند. معمولاً افراد با تالاسمی مینور کم خون نیستند اما ممکن است زنان مبتلا، در زمان حاملگی دچار کم خونی شوند . به طور کلی افراد با تالاسمی مینور بیمار نیستند و بسیاری از آنها از اینکه خونشان با افراد عادی تفاوت دارد بی اطلاع هستند. افراد با تالاسمی مینور را ناقل نیز می نامند و این حالت زمانی خطرناک است که دو فرد مبتلا به تالاسمی مینور (ناقل) با یکدیگر ازدواج نمایند که در این صورت احتمال دارد که فرزندان آینده آنها به تالاسمی ماژور مبتلا شوند. تالاسمی ماژور تالاسمی ماژور یک بیماری جدی است که فرزند مبتلا، دو ژن معیوب را از پدر و مادر به ارث برده است. در این نوع تالاسمی، اختلال خونی شدید است به طوری که کودکان مبتلا نیازمند دریافت خون و سایر درمان های طبی خواهند بود . بچه های مبتلا به تالاسمی ماژور در هنگام تولد سالم هستند اما بین 3 تا 18 ماهگی ممکن است دچار کم خونی شوند. طبقه بندی ژنتیکی: دو نوع اصلی تالاسمی، نوع آلفا و بتا می باشد و براساس نام دو زنجیره ی پروتئینی که هموگلوبین نرمال می سازند آلفا و بتا نامگذاری شده اند. تالاسمی آلفا و بتا، هر دو نوع وخیم و خفیف دارند . آلفا تالاسمی تالاسمی آلفا به علت کاهش میزان زنجیره آلفا ایجاد می شود . تالاسمی آلفا به طور شایع در آفریقا، خاورمیانه، هند، آسیای جنوبی، جنوب چین و نواحی مدیترانه ای یافت میشود . این بیماری به صورت اتوزومال مغلوب به ارث می رسد . خوشه ی ژنی آلفاگلوبولین درانتهای بازوی کوتاه کروموزم 16 قراردارد. در داخل این خوشه ی ژنی دو ژن آلفا یک و آلفا دو قرار دارند که تولیدکننده زنجیره ی آلفا هستند . به دلیل وجود تشابه 95 درصدی ژن های آلفا یک و آلفا دو احتمال جفت شدن نابجای این دو توالی و در نتیجه حذف ژنی وجود دارد. حذف بخشی از ژن آلفاگلوبولین در بیش از 90 درصد موارد عامل ایجاد کننده بیماری است . در 10 درصد موارد نیز حذف های کوچک یا موتاسیون های نقطه ای باعث به وجود آمدن علائم بیماری می شوند. بتا تالاسمی تالاسمی بتا در اثر نقص در تولید زنجیره بتا ایجاد می شود . این بیماری در مردم نواحی مدیترانه نظیر یونان و ایتالیا ، ایران ، شبه جزیره عربستان ، آفریقا ، جنوب آسیا و جنوب چین یافت میشود . وراثت این بیماری به صورت اتوزومال مغلوب است و در نتیجه والدین ناقل به احتمال 25 درصد ممکن است صاحب فرزند بیمار شوند . تشخیص پیش از تولد تالاسمی بتا در دو مرحله تشخیص ناقلین (مرحله اول) و تشخیص پیش از تولد ( مرحله دوم ) انجام می گیرد . خوشه ی ژنی بتا تالاسمی روی کروموزم 11 قرار دارد . در این خوشه ی ژنی علاوه بر ژن بتاگلوبولین ژنهای دلتا (δ) ، گاما (Gγ, Aγ) و اپسیلون (ε) نیز قرار دارد . این ژنها در مراحل مختلف جنینی بروز مییابند که ژن بتا در بزرگسالان بروز میکند و افراد نرمال دو نسخه از آن را دارند . شدت بیماری بستگی به نوع جهشی که در افراد اتفاق می افتد دارد ، به طورمثال بعضی از جهشهای ژن بتا باعث میشود تولید پروتئین صورت نگرفته یا پروتئین غیر فعال ایجاد میشود . (β0) و برخی جهشها که در نواحی تنظیمی ژن اتفاق میافتد ، منجر به کاهش میزان تولید این پروتئین میشوند .(β+) افراد ناقل که حاملان نقص بتا تالاسمی هستند به دو صورت β/β0 وβ/β+ دیده میشوند . بیماران دارای ژنوتیپ β+/β+ فنوتیپ حد واسط نشان میدهند که این افراد معمولاً زندگی نرمال دارند اما بسته به شدت کم خونی در مواردی چون بیماری و یا بارداری نیازمند دریافت خون میشوند . افراد ماژور β0/β0 وβ0/β+ کم خونی شدید دارند ، گلبولهای قرمز این افراد ناکارآمد هستند . تولید خون در مغز استخوان های این افراد شدت مییابد که این امر در دراز مدت باعث تغییر شکل استخوانها به ویژه استخوانهای پهن میشود ، همچنین این بیماران نیاز به دریافت خون دارند که بیماران را با عوارضی چون افزایش میزان آهن روبهرو میکند و به دلیل کم خونی و بالا رفتن آهن بدن نارسایی قلبی و آریتمی (ناهماهنگی ضربان قلب) نیز ایجاد می شود و هم چنین پوست زرد رنگ ، تغییرات در کبد ، طحال و مثانه دیده میشود . اگر دو فرد حامل تالاسمی بتا با یکدیگر ازدواج کنند یکی از سه مورد زیر اتفاق خواهد افتاد : نوزاد ممکن است دو ژن نرمال دریافت کند (از هرکدام از والدین یک ژن) و خون نرمال و سالم داشته باشد (احتمال %25) ، نوزاد ممکن است یک ژن نرمال از یکی از والدین و یک ژن ناقص از والد دیگر دریافت کند و حامل تالاسمی شود (احتمال %50) ، نوزاد ممکن است هر دو ژن را ژن تالاسمی دریافت کند (از هر کدام از والدها یکی) و به تالاسمی خفیف تا شدید مبتلا شود (احتمال %25) تشخیص تالاسمی تشخیص تالاسمی معمولاً با آزمایش خون شامل شمارش کامل خون، آزمایشهای ویژه هموگلوبین و آزمایشهای ژنتیکی صورت میگیرد. علائم تالاسمی خستگی، ضعف و بی حالی، رنگ پریدگی یا پوست زرد رنگ، بدشکلی استخوان صورت، رشد کند و آهسته، تورم شکم و ادرار تیره می باشد. هرچند علائم تالاسمی به نوع و شدت بیماری بستگی دارد. برخی نوزادان علائم تالاسمی را در زمان تولد نشان میدهند درحالیکه علائم این بیماری در برخی نوزادان در 2 سالگی بروز پیدا میکند. برخی از افرادی که فقط دارای یک ژن هموگلوبین معیوب هستند، هیچ گونه علائم تالاسمی را نشان نمیدهند. تالاسمی از طریق آزمایش کامل خون(CBC) و تحقیقات خاصی بر روی هموگلوبین فرد تشخیص داده میشود CBC .اطلاعات مربوط به میزان هموگلوبین و انواع مختلف گلبول های خون ، مثل گلبول های قرمز خون ، را از طریق نمونه خون در دسترس قرار می دهد . تعداد گلبول های قرمز خون و هموگلوبین در افراد مبتلا به تالاسمی کمتر از حد نرمال است . تنها نشانه در حاملان بیماری تالاسمی ، ممکن است گلبوهای قرمز آنها کمی کوچکتر از حد معمول باشند . تحقیقات هموگلوبین ، نوع هموگلوبین افراد را در نمونه ی خون می سنجد. تالاسمی به خاطر علائم و نشانه های آن از قبیل آنمی شدید ، معمولاً در اوایل کودکی تشخیص داده می شود . در برخی از افرادی که مبتلا به انواع خفیف تری از تالاسمی هستند ، پس از یک آزمایش خون معمولی ، کم خونی آنها تشخیص داده می شود . اگر کودکی مبتلا به کم خونی باشد ، پزشکان به تالاسمی مشکوک خواهند شد و او را جزء افراد در خطر ابتلا به تالاسمی قرار خواهند داد ، برای تشخیص کم خونی که از کمبود آهن ایجاد می شود و کم خونی ایجاد شده توسط تالاسمی ، آزمایش سنجش میزان آهن خون انجام می گیرد ، آنمی کمبود آهن به این علت اتفاق می افتد ، که بدن آهن کافی برای ساختن هموگلوبین ندارد اما کم خونی در تالاسمی به خاطر کمبود آهن صورت نمی گیرد و به خاطر مشکل زنجیره های گلوبین آلفا یا بتا است . مکمل های آهن هیچ تأثیری در بهبودی کم خونی ناشی از تالاسمی ندارند ، به خاطر اینکه کمبود آهن مشکل کار نیست. بررسی ژنتیکی خانواده ها برای تشخیص تالاسمی بسیار مؤثر است . در این روش سابقه ی خانوادگی مورد بررسی قرار می گیرد و هر یک از اعضاء خانواده مورد آزمایش خون و بررسی ژنتیکی قرار می گیرند. درمان تالاسمی درمان تالاسمی به نوع و شدت بیماری بستگی دارد افراد حامل بیماری ، معمولاً هیچ علائم یا نشانه ای ندارند و هیچ درمانی نیز نیاز ندارند .افراد مبتلا به نوع متوسط بیماری (مثل تالاسمی اینترمدیا) گاهی به تزریق خون نیاز دارند ، مثلاً زمان هایی که به خاطر یک بیماری دچار استرس می شوند . اگر بیماری فردی که مبتلا به تالاسمی اینترمدیا است ، وخیم تر شود و نیاز به تزریق های خون مکررتری پیدا کند ، آن فرد دیگر ، تالاسمی اینترمدیا ندارد و به تالاسمی ماژور مبتلا شده است . قابل ذکر است که تالاسمی اینترمدیا نوعی از تالاسمی می باشد که علایمی ما بین تالاسمی مینور و ماژور را از خود نشان می دهد . افراد مبتلا به تالاسمی های شدیدتر ، بیماری جدی و خطرناک دارند ، این بیماران با تزریقات مکرر خون ، درمان شلاته آهن و پیوند مغزاستخوان مورد درمان قرار می گیرند ، اگر این افراد مورد درمان قرار نگیرند در اوایل سنین کودکی جان خود را از دست می دهند . افرادی که درمان های خود را با موفقیت انجام دهند ، می توانند تا سنین بالاتری عمر کنند اگر تالاسمی در خانواده شما وجود دارد، پزشک متخصص ژنتیک می تواند به شما کمک کند که چه آزمایشهای ژنتیکی برای شما و سایر اعضاء خانواده شما مناسب میباشد. همینطور اگر قصد ازدواج داشته باشید می توانید از طریق مشاوره ژنتیک از میزان خطر انتقال این بیماری به فرزندان خود مطلع شوید. برای انجام مشاوره ژنتیک میتوانید به آزمایشگاه پاتوبیولوژی و ژنتیک رصد یا یک آزمایشگاه ژنتیک معتبر مراجعه بفرمایید. علاوهبراین اگر یک زوج ناقل ژنهای معیوب سازنده هموگلوبین باشند میتوانند قبل از بارداری با مراجعه به پزشک متخصص ژنتیک و انجام آزمایشهای ژنتیک، نوع ژن معیوب را مورد شناسایی قرار داده سپس در هفته 10 تا 15 بارداری وضعیت جنین را از لحاظ ابتلا به بیماری مذکور مورد بررسی قرار دهند.
آخرین مطالب

دانستنی ها درباره بیماری تالاسمی
اردیبهشت 21, 1401
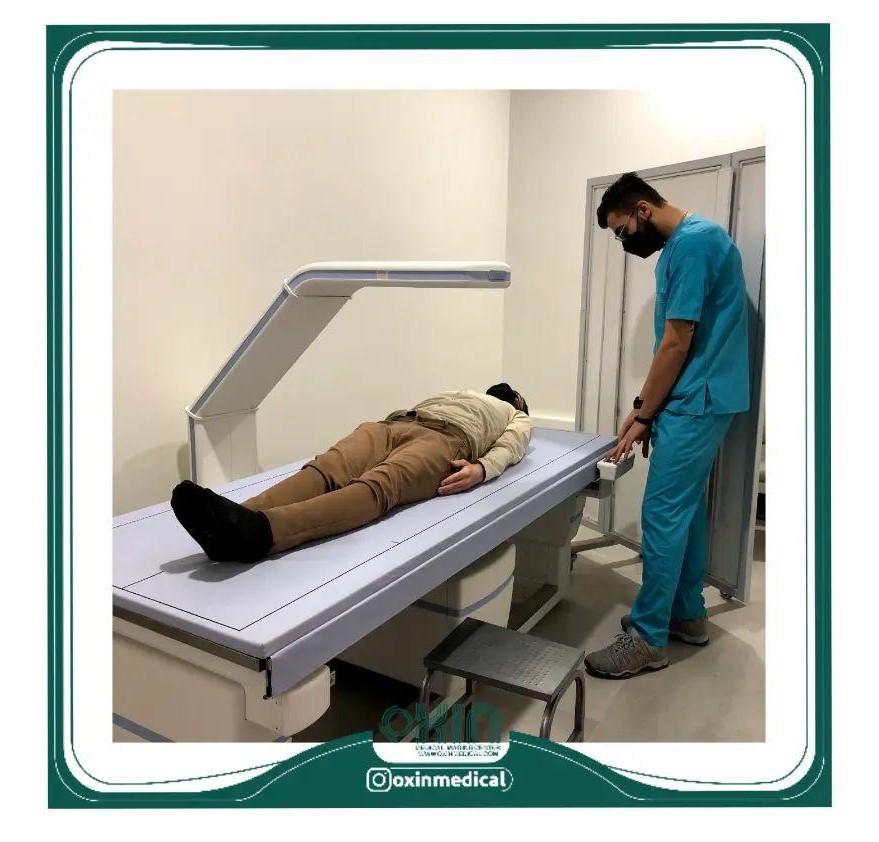
اهمیت سنجش تراکم استخوان
فروردین 27, 1401
ویروس کرونا (CORONA VIRUS)
بهمن 15, 1398

شناسایی 200 بیماری غیرواگیر و ۶۰ عامل خطر مرگومیر
بهمن 05, 1398
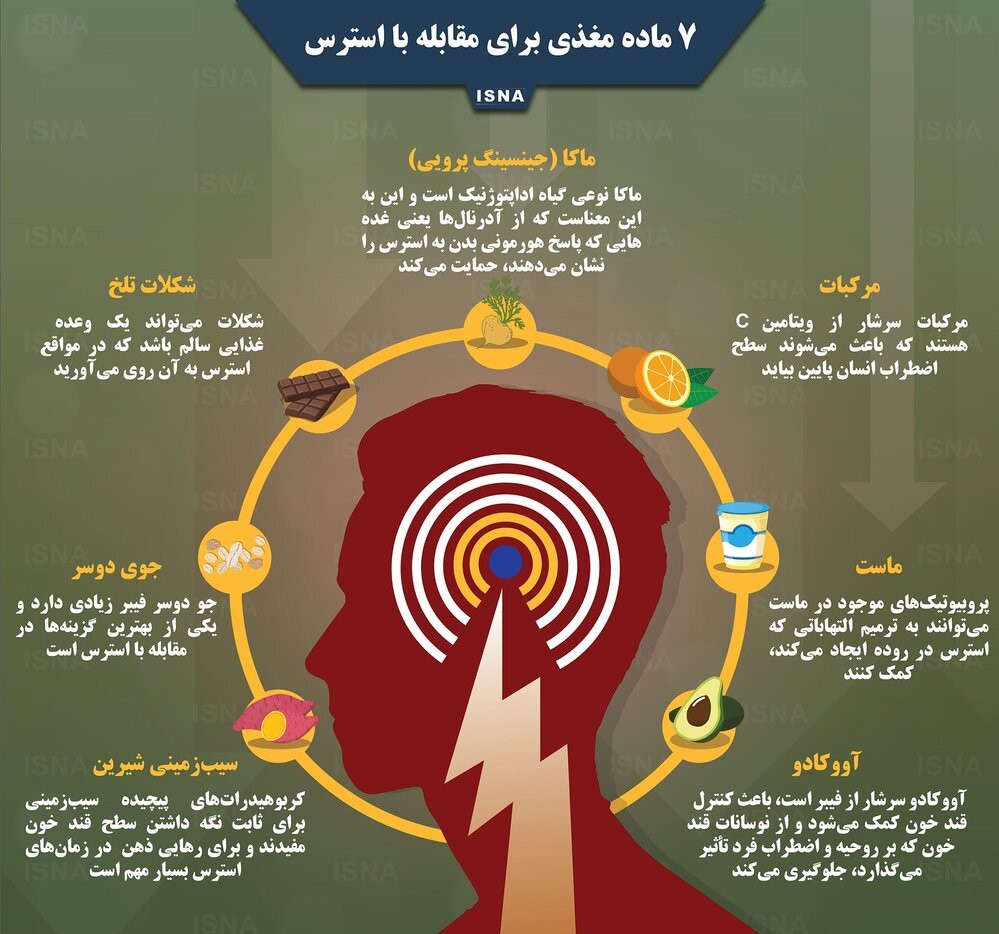
هفت ماده مغذی برای مقابله با استرس
بهمن 05, 1398
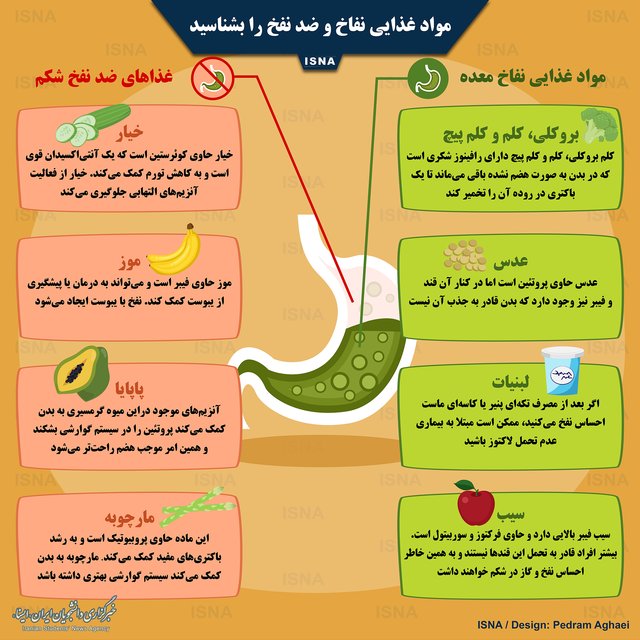
کدام مواد غذایی نفاخ و ضد نفخ هستند
آذر 02, 1398
برای تقویت سیستم ایمنی و رفع ضعف چه واکسنی استفاده کنید؟
آبان 21, 1398

شناسایی بیش از هزار نوع ویروس سرماخوردگی
آبان 19, 1398
آسیب سرخک به حافظه سیستم ایمنی بدن
آبان 11, 1398
برای مسموم نشدن به ایمن سازی توجه کنید
آبان 02, 1398